



New Drug Designations - September 2023
Shots:
-
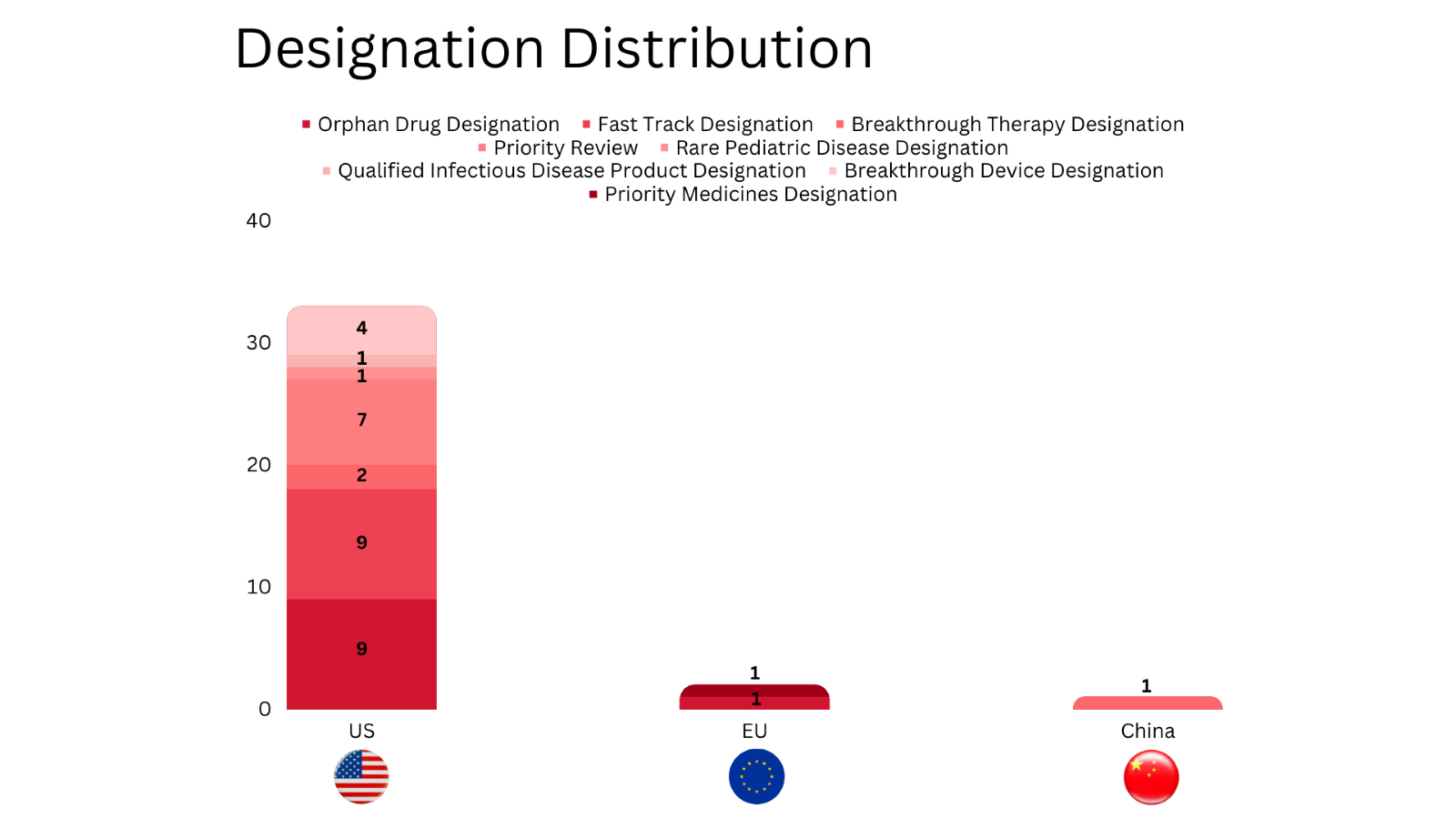
PharmaShots' designation report provides a concise overview of several drugs and their designations by the US FDA, the EU, and China. This month’s report includes 10 biological drugs, 15 small molecules, 5 cell and gene therapies, 4 devices, 1 vaccine and 1 microbiota
-
MicuRx’ Contezolid and Contezolid acefosamil, focused on the treatment of Diabetic Foot Infection (DFI), are the drugs to receive both QIDP and FTD by the US FDA
-
PharmaShots has compiled a list of a total of 32 drugs and 4 devices awarded with designations by multiple regulatory bodies in Sep 2023

INT230-6

-
The ODD has been granted to the formulation of INT230-6 consisting of cisplatin, vinblastine sulfate, and the diffusion enhancer SHAO-FA [8-((2-hydroxybenzoyl) amino) octanoate]
-
The data from the P-I/II study, presented at ASCO’23, showed the m-OS of INT230-6 alone in refractory STS patients was extended by nearly 450 days with favorable safety. The local administration of INT230-6 demonstrated a systemic immune response in various sarcoma subtypes that are non-immunogenic cancers
-
Intensity anticipates a P-III registration trial of INT230-6 for the treatment of STS
Pitolisant

-
Harmony is currently evaluating the efficacy and safety of pitolisant in the double-blind, PBO-controlled, randomized registrational P-III trial (INTUNE) in adult patients with IH.
-
Topline results are expected in the Q4’23 following enrollment completion 9mos. before planned
Veldona

-
Ainos has conducted the following trials for the treatment of oral warts in HIV-seropositive patients:
-
An open-label pilot study in 15 HIV-seropositive males with multiple oral warts
-
An open-label pilot study in 15 HIV-seropositive males with multiple oral warts
-
A single-blind, dose-ranging study in 21 HIV-seropositive participants with multiple oral warts receiving a combination anti-retroviral therapy with/without PI
-
A double-blind, PBO-controlled study in a total of 80 HIV-seropositive subjects on highly active antiretroviral therapy ("HAART"), randomized to IFN-alpha (n=60) or PBO (n=20) for up to 24wks.
-
-
The trials showed positive therapeutic efficacy and a lack of significant toxicity
-
Ainos intends a pre-IND meeting with the US FDA to develop its Veldona formulation in the P-III clinical studies
Alisertib
-
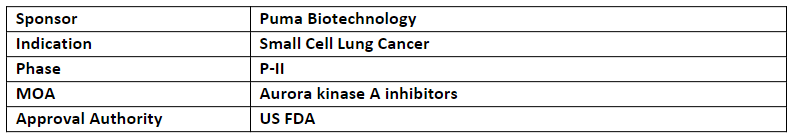
Puma anticipates initiation of P-II (PUMA-ALI-4201) study evaluating alisertib to treat esSCLC in the H2’23. It received IND clearance in Aug’23
-
Alisertib (oral) is a selective, small-molecule, aurora kinase A inhibitor developed for the treatment of patients with small cell lung cancer (SCLC)
BDC-1001

-
The US FDA granted ODD to the company’s BDC-1001 for the treatment of G/GEJ cancer
-
Bolt will initiate two P-II studies evaluating BDC-1001 (Boltbody Immune-Stimulating Antibody Conjugate) for the treatment of patients with HER2-expressing solid tumors in the US, the EU, and South Korea:
-
NCT04278144: for patients with colorectal, endometrial, and gastroesophageal cancers
-
NCT05954143: for patients with breast cancer
Setanaxib

-
Calliditas anticipates conducting a randomized, PBO-controlled P-II trial evaluating setanaxib for Alport syndrome enrolling 20 participants in Q4’23
-
The company’s setanaxib is being investigated in multiple other trials incl. a P-II PoC trial for squamous cell carcinoma of the head and neck (SCCHN), P-IIb trial for primary biliary cholangitis (PBC) and an investigator-led trial for idiopathic pulmonary fibrosis (IPF)
TI-168

-
Following the US FDA’s IND clearance, the company is going to initaite P-I/IIa study of TI-168 for the treatment of Hemophilia A with FVIII inhibitors
-
The company’s TI-168 is next-generation, FVIII specific Treg therapy indicated for Hemophilia A patients with FVIII inhibitors. Further development expected in 2024
NXC-201
-
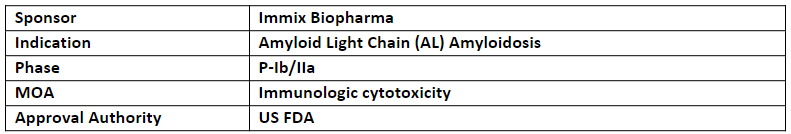
NXC-201 is a next generation CAR-T cell therapy that is being investigated in the P-Ib/IIa study (NEXICART-1)
KB408
-
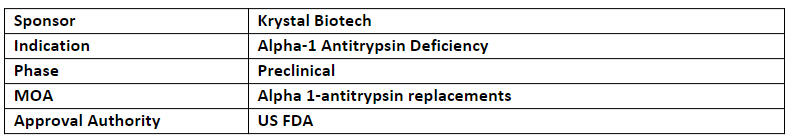
The company’s KB408 is an inhaled (nebulized) formulation of novel replication-defective, non-integrating HSV-1-based vector that delivers two copies of the SERPINA1 transgene, encoding for human alpha-1 antitrypsin protein to treat AATD
MaaT033

-
The ODD has been granted to MaaT033 for improving overall survival in patients undergoing HSCT
-
MaaT033 (capsule) is a donor-derived, high-richness, high-diversity oral Microbiome Ecosystem therapy containing Butycore species designed as an adjunctive and maintenance therapy in allo-HSCT

Contezolid and Contezolid acefosamil

-
The US FDA has also granted QIDP alongwith FTD to the company’s contezolid and contezolid acefosamil for the treatment of diabetic foot infection (DFI)
-
The product has also received QIDP designation and FTD in 2018 to treat acute bacterial skin and skin structure infections (ABSSSI)
-
The global P-III study evaluates contezolid (oral) and contezolid acefosamil (IV) for the treatment of moderate to severe diabetic foot infection (DFI) without concomitant osteomyelitis
Vididencel

-
The US FDA granted FTD based on the promising results demonstrated by vididencel as a monotx. evaluated in the ADVANCE II study for the treatment of AML
-
The company’s vididencel has previously received ODD to treat AML in the US and the EU. Furthermore, Mendus has also received the EMA’s Advanced Therapy Medicinal Product (ATMP) certificate following a review of manufacturing quality and non-clinical data for vididencel
-
The data from the ADVANCE II study in AML, presented at the ASH 2022, showed the ability of vididencel to control MRD and induce durable relapse-free survival in most participants. The company anticipates the next survival update in Q4’23
-
Mendus plans to initiate a pivotal P-II study to assess vididencel + Onureg (azacitidine) in H2’23
-
The vididencel vaccine is based on the company’s proprietary DCOne leukemic cell line and is currently being investigated in AML and ovarian cancer for its anti-tumor activity
Tulmimetostat (CPI-0209)

-
The FTD was granted based on the preclinical data and preliminary clinical results from an ongoing P-I/II trial evaluating tulmimetostat as a monotx. for advanced solid tumors or lymphomas, incl. ARID1A-mutated endometrial and ovarian clear cell carcinoma, DLBCL, peripheral T-cell lymphoma, BAP1-mutated mesothelioma and CRPC. The updated data was presented at the ASCO’23
-
The company’s tulmimetostat is a next-generation investigational targeting EZH1 and 2 to treat advanced, recurrent or metastatic endometrial cancer harboring ARID1A mutations who have progressed on at least one prior line of treatment
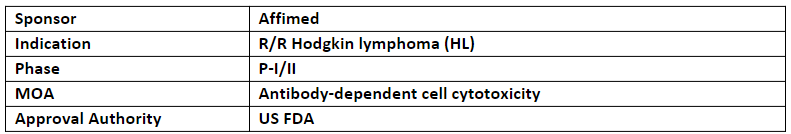
AFM13
-
The P-I/II trial (AFM13-104) evaluating AFM13 in combination with cord blood-derived natural killer cells for the treatment of Hodgkin and non-Hodgkin lymphoma showed
-
ORR of 94%, CR rate of 71% and a well-managed safety profile RP2D
-
In patients with r/r HL (n=31) the ORR was of 97% and CR was of 77%
-
Based on the P-I/II results, Affimed is conducting a P-II (LuminICE-203) evaluating AFM13 + AlloNK which incl. an exploratory cohort of CD30+ peripheral T-cell lymphoma patients
9MW3011 (US: MWTX-003/DISC-3405)

-
9MW3011 is an anti-TMPRSS6 Ab for regulating the level of hepcidin, inhibit the absorption and release of iron, and lower the serum iron level, thus regulating the iron homeostasis in vivo except Polycythemia Vera, 9MW3011 is expected to be assessed in β-thalassemia
-
FDA and NMPA both have approved IND application for 9MW3011 whereas first patient has been dosed in Mar’23 for PV patients
-
DISC Medicine holds exclusive rights to develop and commercialize 9MW3011 in the US, the EU and other territories except Great China and Southeast Asia. Mabwell will receive a total of up to $412.5M of down payment and milestone payments, as well as royalties on net sales
MYTX-011
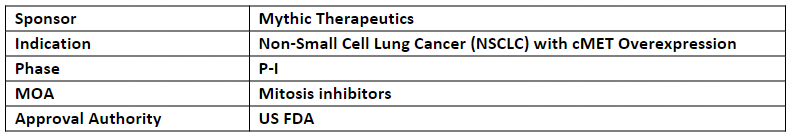
-
Mythic’s MYTX-011 is being investigated in the FIH, open-label, multi-center, dose escalation and dose expansion P-I study (KisMET-01) for the treatment of locally advanced, recurrent or metastatic NSCLC with cMET over expression
-
MYTX-011 is an investigational cMET-targeting ADC based on FateControl technology developed to allow ADCs to navigate inside the cells, potentially increasing delivery of anti-cancer agents to tumor cells
IDE161

-
The US FDA’s FTD has been granted to IDE161 (PARG inhibitor) for the treatment of advanced or metastatic ovarian cancer with germline or somatic BRCA 1/2 mutations who are Pt-resistant and have received prior antiangiogenic and PARP inhibitor therapies
-
Ideaya is investigating IDE161 in the P-I FIH study for its safety, tolerability, PK/PD and preliminary efficacy for the treatment of HERD +ve solid tumors. The P-I expansion part will incl. patients having HRD+ breast cancer, ovarian cancer and other selected solid tumors
-
Additionally, the initial results from the dose escalation cohorts demonstrated tumor shrinkage in multiple HER+ve solid tumor patients, incl. a BRCA 1/2m endometrial cancer. This supported expansion into priority tumor indications while determining the P-II dose
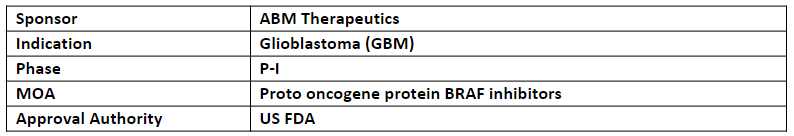
ABM-1310
-
The US FDA has granted FTD to ABM-1310 for the treatment of BRAF V600E mutant Glioblastoma (GBM).
-
In Jul, it has also received ODD by the US FDA for malignant gliomas incl. GBM
KT-333

-
The US FDA has also granted ODD to KT-333 for the treatment of both CTCL and PTCL in 2022
-
The company’s asset, KT-333, is being evaluated in a P-Ia trial for its safety, tolerability, PK/PD and clinical activity dosed weekly for the treatment of adult patients with r/r lymphomas, leukemias and solid tumors
-
KT-333, highly selective degrader of STAT3, is under development to treat multiple STAT3-dependent pathologies, incl. hematological malignancies and solid tumors

Vyvgart (efgartigimod alfa Injection)

-
The BTD was granted based on the data from both global and Chinese patients recruited in the trial (ADHERE)
-
In Jul’23, Zai Lab and argenx highlighted positive topline data from the (ADHERE) trial assessing efgartigimod alfa injection (SC) in adults with CIDP which are as follows:
-
The trial met 1EP (p=0.000039) where 61% reduction (HR: 0.39 95% CI: 0.25; 0.61) in the risk of relapse vs PBO was observed
-
The 67% patients of global trial in open-label Stage A showed evidence of clinical improvement (ECI) indicating role of IgG autoantibodies in the underlying biology of CIDP
-
The drug showed consistent safety and tolerability profile
-
-
The results of the subgroup analysis for (ADHERE) trial patients in China were consistent with global results:
-
The reduction of 69% in relapse risk with efgartigimod SC vs PBO was observed
-
The mainland Chinese patients demonstrated a similar level of response compared to the global population, with 78% of the mainland Chinese participants treated with open label efgartigimod SC demonstrating confirmed ECI
-
The efgartigimod SC’s safety profile was consistent in the mainland Chinese subgroup, with that observed in the global population
-
Pegozafermin

-
The US FDA granted BTD based on the results from the P-IIb (ENLIVEN) study evaluating pegozafermin in patients with NASH.
-
The results showed, the groups receiving 44mg q2w and 30mg qw met the 1EPs as per the FDA guidance and demonstrated improvements in liver fat, non-invasive markers of liver fibrosis and inflammation, and in other metabolic and lipid markers
-
Furthermore, the descriptive analysis of biopsy confirmed F4 patients showed 45% of those treated with pegozafermin experienced at least one-stage improvement in liver fibrosis without any worsening of NASH by week 24 vs sole patient who received a PBO. Pegozafermin was well-tolerated, and its safety profile was consistent with previous studies
-
Pegozafermin is a specifically engineered glycopegylated analog of FGF21 designed for the treatment of non-alcoholic steatohepatitis (NASH) and severe hypertriglyceridemia (SHTG)
INO-3107

-
The BTD has been granted based on the results from completed open-label, multicenter P-I/II study evaluating INO-3107 in patients with HPV-6 and/or HPV-11-related RRP.
-
In the trial, 81.3% of patients (26/32) saw a reduction in surgical interventions in the year following INO-3107 administration. 28.1% (9/32) required no surgical interventions during or after treatment. Before treatment, patients had a median of 4 surgeries (range 2-8)/ year, which decreased by a median of 3 surgeries after INO-3107 administration. Patients received 4 doses of INO-3107, with any surgery after the first dose counted against efficacy. INO-3107 was well-tolerated by the patients
-
Data from the P-I/II study was presented at ABEA’23 and the European Laryngological Association's Annual Meeting. It was also published in the journal, The Laryngoscope, under the title "Interim Results of a Phase 1/2 Open-Label Study of INO-3107 for HPV-6 and/or HPV-11–Associated RRP"
-
INOVIO anticipates a pivotal study of INO-3107 in Q1’24, subject to FDA clearance under CRO, KOL and investigators for the treatment of RRP
-
INO-3107 is an investigational DNA therapeutic candidate indicated to bring out a targeted T cell response against HPV-6 and HPV-11. This BTD follows receipt of EC’s and the US FDA’s ODD in May 2023 and 2020 respectively

Welireg (belzutifan)

-
The US FDA has accepted and granted priority review for sNDA seeking approval for Welireg (HIF-2α inhibitor) for adult patients with advanced RCC following immune checkpoint and anti-angiogenic therapies. The US FDA’s decision is expected in Jan’24
-
The sNDA was based on the results from the P-III trial (LITESPARK-005) evaluating Welireg (120mg, qd) vs everolimus (10mg, qd) in 746 patients which showed a significant clinical improvement in PFS based on a pre-specified interim analysis along with improvement in 2EPs of ORR
-
Welireg, the first HIF-2α inhibitor therapy approved in the US and is currently approved for adult patients with VHL disease
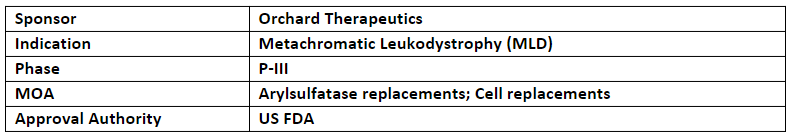
OTL-200
-
The US FDA accepted and granted priority review for BLA of OTL-200 to treat metachromatic leukodystrophy (MLD). The US FDA’s decision is expected in 18th Mar’24 (PDUFA date)
-
Orchard’s OTL-200 has also received both RPDD and RMAT designations from the US FDA and is approved as Libmeldy (atidarsagene autotemcel) by the EC and MHRA
Resmetirom

-
The US FDA accepted the NDA and granted the priority review of NDA for the treatment of patients with NASH with liver fibrosis. The decision is expected in Mar’24
-
The NDA was based on the data from 18 clinicals trials (P-I studies: 12, P-II: 2, & P-III: 4)
-
The company's resmetirom is currently being evaluated in 4 P-III trials incl. MAESTRO-NASH, MAESTRO-NAFLD-1, MAESTRO-NAFLD-OLE and MAESTRO-NASH-OUTCOMES
-
Resmetirom is an oral, thyroid hormone receptor (THR)-β selective agonist developed to target underlying causes of NASH in the liver
Keytruda (pembrolizumab)

-
The US FDA has accepted and granted priority review for sBLA of Keytruda + EBRT + concurrent CT, followed by brachytherapy (concurrent chemoradiotherapy) for the treatment of patients with high-risk locally advanced cervical cancer. The FDA’s decision is expected in Jan’24
-
The sBLA was based on results from the study (KEYNOTE-A18) also known as ENGOT-cx11/GOG-3047 evaluating Keytruda + concurrent chemoradiotherapy that showed a significant improvement in PFS vs concurrent chemoradiotherapy alone
Dupixent (dupilumab)

-
The US FDA has accepted and granted priority review for sBLA of Dupixent for the treatment of children aged 1-11 years with EoE. The decision is expected in Jan’24
-
The sBLA was based on the data from P-III study (EoE KIDS) evaluating the efficacy and safety of Dupixent in children aged 1 to 11 with EoE
-
The results of Part A showed that 1EP was met for the proportion of patients achieving histological disease remission at 16 wks. for tiered dosing regimens based on body weight vs PBO
-
The results of Part B, an active treatment extension period evaluating Dupixent for an additional 36 wks., showed that it met the 2EP of histologic remission for 52 wks.
-
Dupixent also led to increase in body weight for age percentile, an exploratory endpoint in Part A and a 2EP in Part B
Odronextamab

-
The US FDA has accepted and granted priority review for BLA of odronextamab for the treatment of adult patients with r/r FL or r/r DLBCL, who have progressed after at least two prior systemic therapies. The decision is expected in Mar’24
-
The BLA was based on the results of P-I (ELM-1) and pivotal P-II studies (ELM-2) evaluating odronextamab in both FL and DLBCL and were presented at the ASH’23
-
The P-I (ELM-1) trial is an ongoing, open-label, multicenter study to evaluate the safety and tolerability of odronextamab in patients with CD20+ B-cell malignancies previously treated with CD20-directed antibody therapy, incl. an expansion cohort evaluating DLBCL patients who had progressed on CAR-T therapy (post-CAR-T)
-
The P-II (ELM-2) trial is an ongoing, open-label, multicenter pivotal study to evaluate odronextamab in 375 having 5 disease-specific cohorts, incl. DLBCL, FL, mantle cell lymphoma, marginal zone lymphoma and other subtypes of B-NHL
-
Regeneron plans to conduct a broad P-III trial to investigate odronextamab in earlier lines of therapy and other B-NHLs, representing one of the largest clinical programs in lymphoma
-
Odronextamab is an investigational bispecific Ab targeting CD20xCD3 to bridge CD20 on cancer cells with CD3-expressing T cells to facilitate local T-cell activation and cancer-cell killing
Sotatercept

-
The US FDA has accepted and granted priority review for BLA of sotatercept to treat adult patients with PAH. The decision is expected in Mar’24
-
The BLA was based on the results of P-III study (STELLAR) evaluating sotatercept that showed a significant improvement in 6-minute walk distance (6MWD) and eight of nine 2EPs. The results were presented at ACC.23/WCC and published in The New England Journal of Medicine
-
The randomized, double-blind, PBO-controlled, multicenter, parallel-group P-III (STELLAR) trial is to investigate the safety and efficacy of sotatercept in adult patients with PAH (WHO Group 1) being treated with background therapy with WHOFC II/III.

Zatolmilast (BPN14770)

-
The two-way crossover P-II study of zatolmilast in 30 adult males with FXS, where 1EP was safety, an exploratory analysis showed potential efficacy in improving cognition, especially in language domains such as picture vocabulary and oral reading recognition. Additionally, clinically meaningful enhancements in daily functioning were observed
-
The company is currently evaluating zatolmilast in a pivotal P-IIb/III trial which comprises of two randomized, double-blind, PBO-controlled studies of 150 patients each:
-
The zatolmilast clinical program also incl. an OLE trial, Study 302, available to participants after completing Study 204 or Study 301
-
The 1EPs of the trials incl. a cognitive assessment of the efficacy of zatolmilast measured by the cognition crystallized composite score of the NIH Toolbox Cognitive Battery, a calculated score from a series of tests to assess the subjects’ change from baseline of the picture vocabulary and oral reading domains of the NIH-TCB, as well as determinations of the drug’s safety and tolerability while the 2EPs incl. assessments of daily living, caregiver and clinician improvement scales and other domains from the NIH-TCB
-
Zatolmilast is an investigational drug targeting cyclic AMP (cAMP) modulation which promotes the maturation of connections between neurons that are impaired in individuals with FXS

Contezolid and Contezolid acefosamil

-
The US FDA has also granted FTD alongwith QIDP to the company’s contezolid and contezolid acefosamil for the treatment of diabetic foot infection (DFI)
-
The product has also received QIDP designation and FTD in 2018 to treat acute bacterial skin and skin structure infections (ABSSSI)
-
The global P-III study evaluates contezolid (oral) and contezolid acefosamil (IV) for the treatment of moderate to severe diabetic foot infection (DFI) without concomitant osteomyelitis

Selective Cytopheretic Device

-
The breakthrough device submission incl. the preclinical results and the data from FIH study in which the 1EP was met without SCD-related SAEs
-
Selective Cytopheretic Device (SCD) is the company’s patented, FIC, cell-directed device for the use in patients with acute or chronic systolic heart failure and worsening renal function due to cardiorenal syndrome or right ventricular dysfunction awaiting implantation of a left ventricular assist device (LVAD)
Intelligent Network Modulation System

-
The INBRAIN system utilizes the capabilities of graphene. Through their neural platform technology, the company achieves ultra-high signal resolution and employs ML software to interpret therapy-specific biomarkers for delivering highly focused, adaptive neuroelectronic therapy that re-balances pathological neural networks
-
INBRAIN’s Intelligent Network Modulation System is being used as an adjunctive therapy for Parkinson’s patients
Obstructive Hydrocephalus Software Tool

-
The company’s obstructive hydrocephalus software tool has received the US FDA’s 510(k) clearance along with BDD for obstructive hydrocephalus
-
Annalise.ai software tool allows both passive and active notifications for suspected OHCP cases identified on unenhanced head CT scans
Aprevo Technology

-
Carlsmed expects to launch its aprevo device for the treatment of cervical spine disease in the US in 2025
-
This is Carlsmed 2nd BDD. Aprevo is a bespoke devices and are 3D printed and directly handed over to the hospitals for surgery making ensuring sterility and specificity

Iopofosine I 131

-
The company’s iopofosine I 131 has previously received the US FDA’s FTD for the treatment of WM patients who received two or more prior treatment regimens, as well as r/r multiple myeloma and r/r DLBCL
-
Cellectar anticipates topline data from the ongoing P-IIb study of iopofosine I 131 to treat Waldenstrom’s Macroglobulinemia (WM) in Q4’23 and expecting an FDA Priority Review and approval in 2024
Related Post: https://pharmashots.com/16059/new-drug-designations-august-2023
Tags

Disha was a content writer at PharmaShots. She is passionate and curious about recent updates and developments in MedTech and Pharma industry. She covers news related to clinical trial results and updates. She can be contacted at connect@pharmashots.com.










